
新聞中心
NEWS

生信篇 | Unicycler:混合測序時代的細菌基因組組裝利器
Unicycler 是一款專為細菌基因組設計的混合組裝工具,由 Ryan Wick 博士團隊開發。它巧妙結合了二代測序中短讀長(Illumina)的高準確性與三代測序中長讀長(PacBio/Oxford Nanopore)的跨重復優勢,特別適用于高重復、高雜合或復雜結構的微生物基因組組裝。同時,它也是一款可以獨立完成二代測序基因組組裝、三代測序基因組組裝以及二三代測序數據混合組裝的優秀生信軟件,下文將著重介紹二三代測序數據混合組裝。
相較于傳統組裝工具(如SPAdes、IDBA-UD),Unicycler 在解決細菌基因組中重復序列和質粒環狀結構時表現卓越,尤其適合單菌分離樣本或宏基因組分箱后的精細化組裝。

軟件功能亮點
1、混合組裝引擎
短讀長糾錯:利用Illumina數據校正長讀長的測序錯誤,提升組裝準確性。
長讀長橋接:通過PacBio/Nanopore長讀長跨越重復區域,連接短讀長無法覆蓋的斷裂區域。
2、自動化流程整合
內置Bowtie2比對與Pilon糾錯,支持從原始數據到最終環化基因組的全流程自動化。
3、環狀結構識別
自動檢測染色體和質粒的環狀結構,生成完整的閉環序列(若數據支持)。
4、靈活輸入支持
兼容Illumina雙端測序、PacBio CLR/CCS及Nanopore數據,適應不同實驗設計需求。

算法核心解析
Unicycler 的算法設計融合了De Bruijn圖與字符串圖(String Graph)的優勢,分三階段實現高效組裝:
1、短讀長糾錯與初步組裝
De Bruijn圖構建:將短讀長拆分為k-mer,構建圖結構,通過尋找歐拉路徑生成初始Contig。
錯誤剔除:基于k-mer頻率和一致性過濾低覆蓋分支,避免測序噪聲干擾。
2、長讀長進行的圖優化
長讀長比對與糾錯:將長讀長比對至初始Contig,校正其測序錯誤并填充缺口。
字符串圖構建:基于長讀長的重疊關系構建字符串圖,解決重復區域的路徑歧義。
3、路徑選擇與環化處理
啟發式搜索最優路徑:結合讀長覆蓋度和拓撲結構選擇最可能的路徑,生成線性或環狀Scaffold。
自動環化檢測:通過比對末端重疊識別環狀結構,輸出完整染色體/質粒序列。

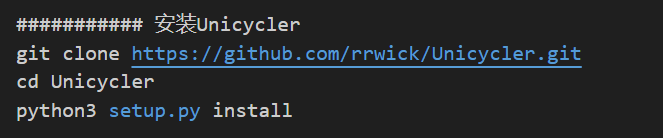
軟件安裝
Github官網上有提供相應源碼,可直接下載安裝,下面提供一種安裝方案,可以安裝最新版的Unicycler。

實戰示例
案例背景:對一株耐藥性的大腸桿菌進行基因組組裝,數據源包含illumina雙端測序(150bp)數據和Nanopore長讀長(N50=15kb)數據。
1、運行命令

? -1/-2:illumina 雙端數據
? - l:長讀長數據
? - o:輸出目錄
2、輸出結果文件
? assembly.fasta: 最終組裝序列
? assembly.gfa: 組裝圖文件,可用Bandage軟件來可視化
? unicycler.log:詳細日志文件,可用于調試與性能評估

結果解讀與優化策略
1、評估指標
Contig N50:若N50接近基因組預期大小(如大腸桿菌~4.6Mb),表明組裝連貫性高。
環化比例:理想情況下,主染色體和質粒應標記為環狀。
BUSCO完整性:使用細菌通用單拷貝基因集評估基因區域的完整性(目標>95%)。
2、常見問題與對策
碎片化Contig:增加長讀長數據中的覆蓋度或調整--min_kmer_coverage參數。
環化失敗:檢查長讀長是否跨越重復區域,或手動使用Circlator等工具進行補環。
嵌合體污染:結合參考基因組比對或基于覆蓋度差異篩選異常區域進行后續分析。

結語
Unicycler 憑借其混合算法與自動化設計,已成為微生物基因組研究的標桿工具。無論是臨床病原體溯源還是環境微生物挖掘,它都能提供高完整度、低錯誤率的組裝結果。下一期我們將帶來Unicycler單獨進行二代測序數據或者三代測序數據組裝的詳細介紹,敬請期待!

電話:021-61283010
網址:m.beb6.cn
郵箱:marketing@bio-chain.com
地址:上海市徐匯區虹漕路421號65幢303室
上海柏辰生物科技有限公司 版權所有 嚴禁復制 網站備案號:滬ICP備09099502號-1 滬公網安備 31010402002899號 網站建設:銘心科技
